Assembly genomes with Bowtie2

Assembling
There are multiple software assemblers, some more suitable or with a better performance depending your purpose, in this case we are going to use ~Bowtie2~ a classic assembler that does not consume too much resources, it is convenient tool for rapid adapter trimming, identification and read merging, other feature is that this assembler is supported by .
Load necesary packages
library(BiocManager)
if (!require("R.utils")){
install("R.utils")
}
if (!require("Rbowtie2")){
install("Rbowtie2")
}
library(R.utils)
library(Rbowtie2)
library(Rsamtools)
library(Rsubread)
library(tidyverse)
library(BiocParallel)
Unzip reference genome
Bowtie2 and many other non-de-novo assemblers use a previously assambled genome as reference, usually, when this genome is downloaded from the NCBI you get a compressed .gz file, you can unzip it with unzip() function from R.utils package.
gunzip("~/Documentos/R/R_bioinformatics/cursos_de_bioinformatica_R/curso_bionformatica_online/Bioinformatica_en_casa/2020-03-23/chromosoms/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa.gz",
remove = F,
overwrite = T)
Crete an index to assemble our sequences
Then you create a index that will be used as reference, in this case we are going to name our index as “yeast” at the end of our path.
bowtie2_build("~/Documentos/R/R_bioinformatics/cursos_de_bioinformatica_R/curso_bionformatica_online/Bioinformatica_en_casa/2020-03-23/chromosoms/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa",
bt2Index = "~/Documentos/R/R_bioinformatics/cursos_de_bioinformatica_R/curso_bionformatica_online/Bioinformatica_en_casa/2020-03-23/chromosoms/yeast", overwrite = T)
Unzip multiple fastq.gz files
Before you align your .fastq sequences to the reference, you have to be sure of working with unzipped files, here we are going to use a for loop to unzip all our .fastq.gz files on few lines of code.
First, you create a vector with all the paths to the .fastq files.
paths <- list.files(path = "~/Documentos/R/R_bioinformatics/cursos_de_bioinformatica_R/curso_bionformatica_online/Bioinformatica_en_casa/2020-03-23/fastq/",
full.names = T)
And then you use a for loop.
for (i in paths) {
gunzip(i,remove = F, overwrite = T)
}
Assembling with Bowtie2
Now that you have all the necessary files unzipped, you need to list on different vectors what bowtie2 will need.
index: The path of our index pair1_paths: A list of all the first paired reads. pair2_paths: A list of all the second paired reads. samOutput: A list with all the names that will have our genomes assembled.
library(stringr)
pair1_paths <- list.files(path = "~/Documentos/R/R_bioinformatics/cursos_de_bioinformatica_R/curso_bionformatica_online/Bioinformatica_en_casa/2020-03-23/fastq",
pattern = "1.fastq$",
full.names = T)
pair2_paths <- list.files(path = "~/Documentos/R/R_bioinformatics/cursos_de_bioinformatica_R/curso_bionformatica_online/Bioinformatica_en_casa/2020-03-23/fastq",
pattern = "2.fastq$",
full.names = T)
samOutput <- str_c(str_remove(pair1_paths,
pattern = "\\_\\d.fastq"),".sam")
index = "~/Documentos/R/R_bioinformatics/cursos_de_bioinformatica_R/curso_bionformatica_online/Bioinformatica_en_casa/2020-03-23/chromosoms/yeast"
You can define the number of cores you will use, that depends on your computer.
register(MulticoreParam(7))
auto_Bowtie2 <- function(index,samOutput,seq1,seq2) {
bowtie2(index,
samOutput = samOutput,
seq1 = seq1,
seq2 = seq2,
overwrite = T,
"--threads 7")
}
for (i in 1:length(samOutput)) {
auto_Bowtie2(index = index,
samOutput = samOutput[i],
seq1 = pair1_paths[i],
seq2 = pair2_paths[i])
}
Convert SAM files to BAM format
SAM files are heavy, for that reason we are going to convert them to a binary format (BAM) with asBam() function from Rsamtools package, you can delete SAM files once you get the BAM files.
path_SAMs <- list.files(path = "~/Documentos/R/R_bioinformatics/cursos_de_bioinformatica_R/curso_bionformatica_online/Bioinformatica_en_casa/2020-03-23/fastq", pattern = "sam$", full.names = T)
for (i in 1:length(path_SAMs)) {
asBam(path_SAMs[i],
overwrite = T)
}
This assemblies can be processed with other tools inside or outside of to apply quality controls and visualize it with other tools like IGV.
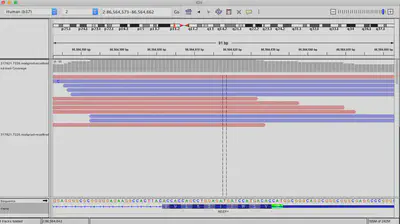
Differential gene expression
If your .fastq files are product of a differential gene expression experiment one of the main steps to find differences is to count the number of reads in each BAM file.
Import BAM files to the R enviroment
You have to list your BAM files in the same way we did it with other files before.
bamfiles <- list.files("~/Documentos/R/R_bioinformatics/cursos_de_bioinformatica_R/curso_bionformatica_online/Bioinformatica_en_casa/2020-03-23/fastq", pattern = ".bam$", full.names = T)
file.exists(bamfiles) #Comprobar que R este leyendo las rutas
## [1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
Also you can check the existence a the nomenclature in our BAM files, this nomenclature have to be de same that the one used in the .gff file with the annotations.
file.exists(bamfiles) #Check their existence
## [1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
bam_files_2 <- BamFileList(bamfiles, yieldSize = 2000000)
seqinfo(bam_files_2[1])# Check the chromosome names
## Seqinfo object with 17 sequences from an unspecified genome:
## seqnames seqlengths isCircular genome
## I 230218 <NA> <NA>
## II 813184 <NA> <NA>
## III 316620 <NA> <NA>
## IV 1531933 <NA> <NA>
## V 576874 <NA> <NA>
## ... ... ... ...
## XIII 924431 <NA> <NA>
## XIV 784333 <NA> <NA>
## XV 1091291 <NA> <NA>
## XVI 948066 <NA> <NA>
## Mito 85779 <NA> <NA>
Count the number of reads
To count the number of reads in each BAM file, an indispensable step to analyze a experiment about differential gene expression, we are going o use featureCounts() from Rsubread to accomplish this task.
featureCount() needs first a list of paths to access to the BAM files, and .gff or .gtf file with the gene coordinates that will be used as benchmarks to find the genes, also you have to precise you are counting genes and you want to get the IDs of those genes, other important stuff it is we have to precise that we are working with paired end reads, in this case, we are going to work with 7 cores and take in consideration only the reads with alignments with quality scores over 20 and we are going to require both ends mapped to count a read.
data <- featureCounts(
files = bamfiles,
annot.ext = "~/Documentos/R/R_bioinformatics/cursos_de_bioinformatica_R/curso_bionformatica_online/Bioinformatica_en_casa/2020-03-23/genes/Saccharomyces_cerevisiae.R64-1-1.99.gff3",
isGTFAnnotationFile = T,
GTF.featureType = "gene",
GTF.attrType = "ID",
isPairedEnd = T,
requireBothEndsMapped = T,
minMQS = 20,
strandSpecific = 2,
nthreads = 7)
##
## ========== _____ _ _ ____ _____ ______ _____
## ===== / ____| | | | _ \| __ \| ____| /\ | __ \
## ===== | (___ | | | | |_) | |__) | |__ / \ | | | |
## ==== \___ \| | | | _ <| _ /| __| / /\ \ | | | |
## ==== ____) | |__| | |_) | | \ \| |____ / ____ \| |__| |
## ========== |_____/ \____/|____/|_| \_\______/_/ \_\_____/
## Rsubread 2.10.5
##
## //========================== featureCounts setting ===========================\\
## || ||
## || Input files : 9 BAM files ||
## || ||
## || 1M_SRR9336468.bam ||
## || 1M_SRR9336469.bam ||
## || 1M_SRR9336470.bam ||
## || 1M_SRR9336471.bam ||
## || 1M_SRR9336472.bam ||
## || 1M_SRR9336473.bam ||
## || 1M_SRR9336474.bam ||
## || 1M_SRR9336475.bam ||
## || 1M_SRR9336476.bam ||
## || ||
## || Paired-end : yes ||
## || Count read pairs : yes ||
## || Annotation : Saccharomyces_cerevisiae.R64-1-1.99.gff3 (GTF) ||
## || Dir for temp files : . ||
## || Threads : 7 ||
## || Level : meta-feature level ||
## || Multimapping reads : counted ||
## || Multi-overlapping reads : not counted ||
## || Min overlapping bases : 1 ||
## || ||
## \\============================================================================//
##
## //================================= Running ==================================\\
## || ||
## || Load annotation file Saccharomyces_cerevisiae.R64-1-1.99.gff3 ... ||
## || Features : 6600 ||
## || Meta-features : 6600 ||
## || Chromosomes/contigs : 17 ||
## || ||
## || Process BAM file 1M_SRR9336468.bam... ||
## || Strand specific : reversely stranded ||
## || Paired-end reads are included. ||
## || Total alignments : 1000000 ||
## || Successfully assigned alignments : 785025 (78.5%) ||
## || Running time : 0.13 minutes ||
## || ||
## || Process BAM file 1M_SRR9336469.bam... ||
## || Strand specific : reversely stranded ||
## || Paired-end reads are included. ||
## || Total alignments : 1000000 ||
## || Successfully assigned alignments : 781156 (78.1%) ||
## || Running time : 0.09 minutes ||
## || ||
## || Process BAM file 1M_SRR9336470.bam... ||
## || Strand specific : reversely stranded ||
## || Paired-end reads are included. ||
## || Total alignments : 1000000 ||
## || Successfully assigned alignments : 794108 (79.4%) ||
## || Running time : 0.04 minutes ||
## || ||
## || Process BAM file 1M_SRR9336471.bam... ||
## || Strand specific : reversely stranded ||
## || Paired-end reads are included. ||
## || Total alignments : 1000000 ||
## || Successfully assigned alignments : 797820 (79.8%) ||
## || Running time : 0.04 minutes ||
## || ||
## || Process BAM file 1M_SRR9336472.bam... ||
## || Strand specific : reversely stranded ||
## || Paired-end reads are included. ||
## || Total alignments : 1000000 ||
## || Successfully assigned alignments : 791847 (79.2%) ||
## || Running time : 0.11 minutes ||
## || ||
## || Process BAM file 1M_SRR9336473.bam... ||
## || Strand specific : reversely stranded ||
## || Paired-end reads are included. ||
## || Total alignments : 1000000 ||
## || Successfully assigned alignments : 788537 (78.9%) ||
## || Running time : 0.04 minutes ||
## || ||
## || Process BAM file 1M_SRR9336474.bam... ||
## || Strand specific : reversely stranded ||
## || Paired-end reads are included. ||
## || Total alignments : 1000000 ||
## || Successfully assigned alignments : 793268 (79.3%) ||
## || Running time : 0.04 minutes ||
## || ||
## || Process BAM file 1M_SRR9336475.bam... ||
## || Strand specific : reversely stranded ||
## || Paired-end reads are included. ||
## || Total alignments : 1000000 ||
## || Successfully assigned alignments : 792930 (79.3%) ||
## || Running time : 0.04 minutes ||
## || ||
## || Process BAM file 1M_SRR9336476.bam... ||
## || Strand specific : reversely stranded ||
## || Paired-end reads are included. ||
## || Total alignments : 1000000 ||
## || Successfully assigned alignments : 788970 (78.9%) ||
## || Running time : 0.03 minutes ||
## || ||
## || Write the final count table. ||
## || Write the read assignment summary. ||
## || ||
## \\============================================================================//
And finally we get a dataframe to start our differential gene expression analyzes with other packages like limma, edgeR or DESeq2
raw_counts <- data$counts