Downloading information from NCBI databases

In this lecture we are going to learn how to use RISmed package to find relevant information for a research, first exploring Pubmed database to find interesting articles about certain subject or organism and next we are going to try more complex examples and databases from NCBI.
First thing you have to do is load RISmed package or install it if you have not done this before.
install.packages("RISmed")library(RISmed)
library(tidyverse)## ── Attaching packages ─────────────────────────────────────── tidyverse 1.3.1 ──## ✓ ggplot2 3.3.5 ✓ purrr 0.3.4
## ✓ tibble 3.1.6 ✓ dplyr 1.0.8
## ✓ tidyr 1.2.0 ✓ stringr 1.4.0
## ✓ readr 2.1.2 ✓ forcats 0.5.1## ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
## x dplyr::filter() masks stats::filter()
## x dplyr::lag() masks stats::lag()Suppose that you are interested on find information in scientific articles about a topic such as machine learning applied to bioinformatics for example, to do that we are going to create a “medLine” object with EUtilsSummary() function to get a summary of the results from PubMed database.
machine_learning <- EUtilsSummary("Machine learning bioinformatics[ti]", type = "esearch", db="pubmed",retmax = 5000)Notice the [ti] tag is used with the string query to precise that you want to find from PubMed database scientific articles that have “Machine learning bioinformatics” on their title. On the other hand use retmax argument to trim the number of results.
Now we use EUtilsGet() to download the results previously found with EUtilsSummary()
machine_learning_results <- EUtilsGet(machine_learning,type="efetch", db="pubmed")And finally we can extract the title, autors, DOIs, cites and many other information from articles related to machine learning and bioinformatics
Article_titles <- ArticleTitle(machine_learning_results)
head(Article_titles)## [1] "SWAAT Bioinformatics Workflow for Protein Structure-Based Annotation of ADME Gene Variants."
## [2] "Machine learning and bioinformatics analysis revealed classification and potential treatment strategy in stage 3-4 NSCLC patients."
## [3] "Bioinformatics Methods in Predicting Amyloid Propensity of Peptides and Proteins."
## [4] "Review of bioinformatics in Azheimer's Disease Research."
## [5] "Identification of Prognostic Biomarkers in Papillary Thyroid Cancer and Developing Non-Invasive Diagnostic Models Through Integrated Bioinformatics Analysis."
## [6] "Democratizing bioinformatics through easily accessible software platforms for non-experts in the field."head(AbstractText(machine_learning_results))## [1] "Recent genomic studies have revealed the critical impact of genetic diversity within small population groups in determining the way individuals respond to drugs. One of the biggest challenges is to accurately predict the effect of single nucleotide variants and to get the relevant information that allows for a better functional interpretation of genetic data. Different conformational scenarios upon the changing in amino acid sequences of pharmacologically important proteins might impact their stability and plasticity, which in turn might alter the interaction with the drug. Current sequence-based annotation methods have limited power to access this type of information. Motivated by these calls, we have developed the Structural Workflow for Annotating ADME Targets (SWAAT) that allows for the prediction of the variant effect based on structural properties. SWAAT annotates a panel of 36 ADME genes including 22 out of the 23 clinically important members identified by the PharmVar consortium. The workflow consists of a set of Python codes of which the execution is managed within Nextflow to annotate coding variants based on 37 criteria. SWAAT also includes an auxiliary workflow allowing a versatile use for genes other than ADME members. Our tool also includes a machine learning random forest binary classifier that showed an accuracy of 73%. Moreover, SWAAT outperformed six commonly used sequence-based variant prediction tools (PROVEAN, SIFT, PolyPhen-2, CADD, MetaSVM, and FATHMM) in terms of sensitivity and has comparable specificity. SWAAT is available as an open-source tool."
## [2] "Precision medicine has increased the accuracy of cancer diagnosis and treatment, especially in the era of cancer immunotherapy. Despite recent advances in cancer immunotherapy, the overall survival rate of advanced NSCLC patients remains low. A better classification in advanced NSCLC is important for developing more effective treatments. The calculation of abundances of tumor-infiltrating immune cells (TIICs) was conducted using Cell-type Identification By Estimating Relative Subsets Of RNA Transcripts (CIBERSORT), xCell (xCELL), Tumor IMmune Estimation Resource (TIMER), Estimate the Proportion of Immune and Cancer cells (EPIC), and Microenvironment Cell Populations-counter (MCP-counter). K-means clustering was used to classify patients, and four machine learning methods (SVM, Randomforest, Adaboost, Xgboost) were used to build the classifiers. Multi-omics datasets (including transcriptomics, DNA methylation, copy number alterations, miRNA profile) and ICI immunotherapy treatment cohorts were obtained from various databases. The drug sensitivity data were derived from PRISM and CTRP databases. In this study, patients with stage 3-4 NSCLC were divided into three clusters according to the abundance of TIICs, and we established classifiers to distinguish these clusters based on different machine learning algorithms (including SVM, RF, Xgboost, and Adaboost). Patients in cluster-2 were found to have a survival advantage and might have a favorable response to immunotherapy. We then constructed an immune-related Poor Prognosis Signature which could successfully predict the advanced NSCLC patient survival, and through epigenetic analysis, we found 3 key molecules (HSPA8, CREB1, RAP1A) which might serve as potential therapeutic targets in cluster-1. In the end, after screening of drug sensitivity data derived from CTRP and PRISM databases, we identified several compounds which might serve as medication for different clusters. Our study has not only depicted the landscape of different clusters of stage 3-4 NSCLC but presented a treatment strategy for patients with advanced NSCLC."
## [3] "Several computational methods have been developed to predict amyloid propensity of a protein or peptide. These bioinformatics tools are time- and cost-saving alternatives to expensive and laborious experimental methods which are used to confirm self-aggregation of a protein. Computational approaches not only allow preselection of reliable candidates for amyloids but, most importantly, are capable of a thorough and informative analysis of a protein, indicating the sequence determinants of protein aggregation, identifying the potential causal mutations and likely mechanisms. Bioinformatics modeling applies several different approaches, which most typically include physicochemical or structure-based modeling, machine learning, or statistics based modeling. Bioinformatics methods typically use the amino acid sequence of a protein as an input, some also include additional information, for example, an available structure. This chapter describes the methods currently used to computationally predict amyloid propensity of a protein or peptide. Since the accuracy of bioinformatics methods may be highly dependent on reference data used to develop and evaluate the predictors, we also briefly present the main databases of amyloids used by the authors of bioinformatics tools."
## [4] "Alzheimer's disease (AD) is a severe neurodegenerative disease with slow course of onset and deterioration with time. With the speedup of global aging, AD has become a disease that seriously threatens the physical health of the elderly; therefore, the effective prevention and treatments of AD is an extremely important area of study for researchers and clinicians. Rapid technological developments have promoted the analysis of various kinds of complex data sets using machine learning methods. The common machine learning algorithms, such as Lasso, SVM and Random Forest, are very important in AD research. To help accelerate AD-related research, we review some recent research progress on Alzheimer's disease, including database, image analysis, gene expression, etc., which can provide AD researchers with more comprehensive research methods."
## [5] "Papillary thyroid cancer (PTC) is the most frequent subtype of thyroid carcinoma, mainly detected in patients with benign thyroid nodules (BTN). Due to the invasiveness of accurate diagnostic tests, there is a need to discover applicable biomarkers for PTC. So, in this study, we aimed to identify the genes associated with prognosis in PTC. Besides, we performed a machine learning tool to develop a non-invasive diagnostic approach for PTC. For the study's purposes, the miRNA dataset GSE130512 was downloaded from the GEO database and then analyzed to identify the common differentially expressed miRNAs in patients with non-metastatic PTC (nm-PTC)/metastatic PTC (m-PTC) compared with BTNs. The SVM was also applied to differentiate patients with PTC from those patients with BTN using the common DEMs. A protein-protein interaction network was also constructed based on the targets of the common DEMs. Next, functional analysis was performed, the hub genes were determined, and survival analysis was then executed. A total of three common miRNAs were found to be differentially expressed among patients with nm-PTC/m-PTC compared with BTNs. In addition, it was established that the autophagosome maturation, ciliary basal body-plasma membrane docking, antigen processing as ubiquitination & proteasome degradation, and class I MHC mediated antigen processing & presentation are associated with the pathogenesis of PTC. Furthermore, it was illustrated that RPS6KB1, CCNT1, SP1, and CHD4 might serve as new potential biomarkers for PTC prognosis. RPS6KB1, CCNT1, SP1, and CHD4 may be considered as new potential biomarkers used for prognostic aims in PTC. However, performing validation tests is inevitable in the future."
## [6] "NA"Also you can create a dataframe to read them.
interesting_articles <- data.frame(Title = head(ArticleTitle(machine_learning_results)),
Abstract = head(AbstractText(machine_learning_results))) %>% as_tibble()
interesting_articles$Title[1]## [1] "SWAAT Bioinformatics Workflow for Protein Structure-Based Annotation of ADME Gene Variants."interesting_articles$Abstract[1]## [1] "Recent genomic studies have revealed the critical impact of genetic diversity within small population groups in determining the way individuals respond to drugs. One of the biggest challenges is to accurately predict the effect of single nucleotide variants and to get the relevant information that allows for a better functional interpretation of genetic data. Different conformational scenarios upon the changing in amino acid sequences of pharmacologically important proteins might impact their stability and plasticity, which in turn might alter the interaction with the drug. Current sequence-based annotation methods have limited power to access this type of information. Motivated by these calls, we have developed the Structural Workflow for Annotating ADME Targets (SWAAT) that allows for the prediction of the variant effect based on structural properties. SWAAT annotates a panel of 36 ADME genes including 22 out of the 23 clinically important members identified by the PharmVar consortium. The workflow consists of a set of Python codes of which the execution is managed within Nextflow to annotate coding variants based on 37 criteria. SWAAT also includes an auxiliary workflow allowing a versatile use for genes other than ADME members. Our tool also includes a machine learning random forest binary classifier that showed an accuracy of 73%. Moreover, SWAAT outperformed six commonly used sequence-based variant prediction tools (PROVEAN, SIFT, PolyPhen-2, CADD, MetaSVM, and FATHMM) in terms of sensitivity and has comparable specificity. SWAAT is available as an open-source tool."Extracting and anallizing data
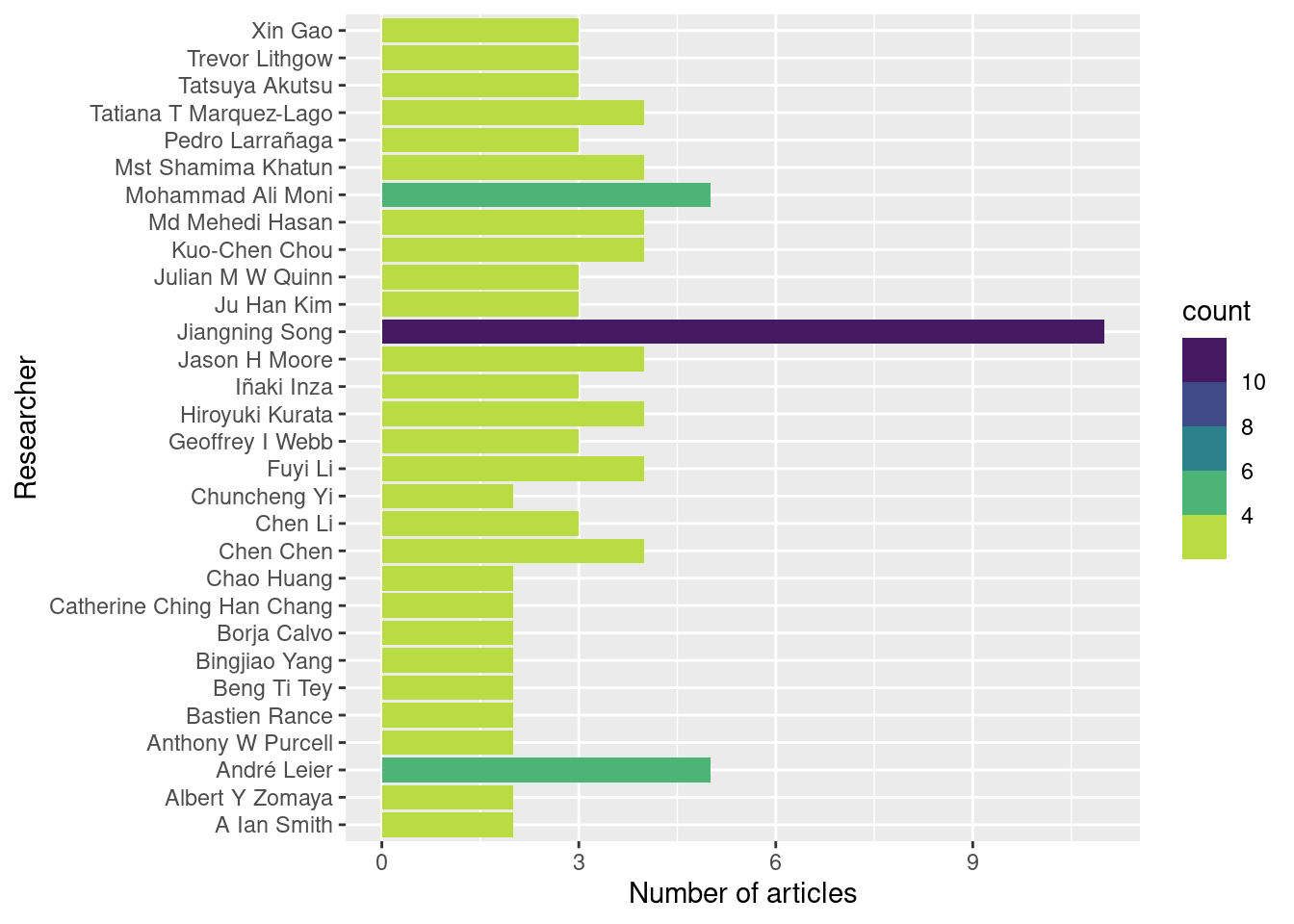
With the obtained information you can do some analyses for example find who are the top researches working on machine learning and bioinformatics or at least those who most publish.
Autors <- Author(machine_learning_results) %>% #Get the Author's name of our 5000 papers
map(., as_tibble) %>% # convert all the dataframes to tibbles
bind_rows() # bind all the dataframes
Autors <- Autors %>% # Get the fullname in a only one string by author
transmute(., complete_name = str_c(Autors$ForeName,
Autors$LastName,
sep = " "))
Autors_summary <- Autors %>%
group_by(.,complete_name) %>% # Group the names
summarize(., count = n()) %>% # count the names to find which researcher have more papers
arrange(., desc(count)) %>% # arrange them by the number of papers
na.omit() %>% # delete NAs
slice_head(., n = 30) #subset the top 30 researcher with more papersFinally you will get a dataframe like this and you can try to plot it with ggplot package
Autors_summary## # A tibble: 30 × 2
## complete_name count
## <chr> <int>
## 1 Jiangning Song 11
## 2 André Leier 5
## 3 Mohammad Ali Moni 5
## 4 Chen Chen 4
## 5 Fuyi Li 4
## 6 Hiroyuki Kurata 4
## 7 Jason H Moore 4
## 8 Kuo-Chen Chou 4
## 9 Md Mehedi Hasan 4
## 10 Mst Shamima Khatun 4
## # … with 20 more rowsggplot(Autors_summary,
aes(complete_name,count, fill = count)) +
geom_col() +
coord_flip()+
scale_fill_viridis_b(direction = -1)+
xlab("Researcher")+
ylab("Number of articles")